inferCNV用与探索肿瘤单细胞RNA-seq数据,分析其中的体细胞大规模染色体拷贝数变化(copy number alterations, CNA), 例如整条染色体或大片段染色体的增加或丢失(gain or deletions)。工作原理是,以一组”正常”细胞作为参考,分析肿瘤基因组上各个位置的基因表达量强度变化. 通过热图的形式展示每条染色体上的基因相对表达量,相对于正常细胞,肿瘤基因组总会过表达或者低表达。
需要准备的数据
- raw counts 表达矩阵,行名是基因,列名是细胞编号
- 细胞的注释文件。一共两列,第一列是细胞编号,第二列是细胞的分组
- 基因位置文件,一共四列,第一列是基因,第二列为染色体信息,第三列为起始位置,第四列为终止位置
脚本
1 | library(argparse) |
脚本使用
1 | /TJPROJ5/SC/software/miniconda3/envs/scrna_py3/bin/Rscript infercnv_argparse_v1.r \ |
注意点:
如果不设置
ref_group_name, 那么inferCNV会使用所有细胞的平均值进行后续分析。关键参数是
cutoff, 用于选择哪些基因会被用于分析(在所有细胞的平均表达量需要大于某个阈值)。这个需要根据具体的测序深度来算,官方教程建议10X设置为0.1,smart-seq设置为1。
文件输出
- infercnv.preliminary.png : 初步的inferCNV展示结果(未经去噪或HMM预测)
- infercnv.png : 最终inferCNV产生的去噪后的热图.
- infercnv.references.txt : 正常细胞矩阵.
- infercnv.observations.txt : 肿瘤细胞矩阵.
- infercnv.observation_groupings.txt : 肿瘤细胞聚类后的分组关系.
- infercnv.observations_dendrogram.txt : NEWICK格式,展示细胞间的层次关系.
执行过程
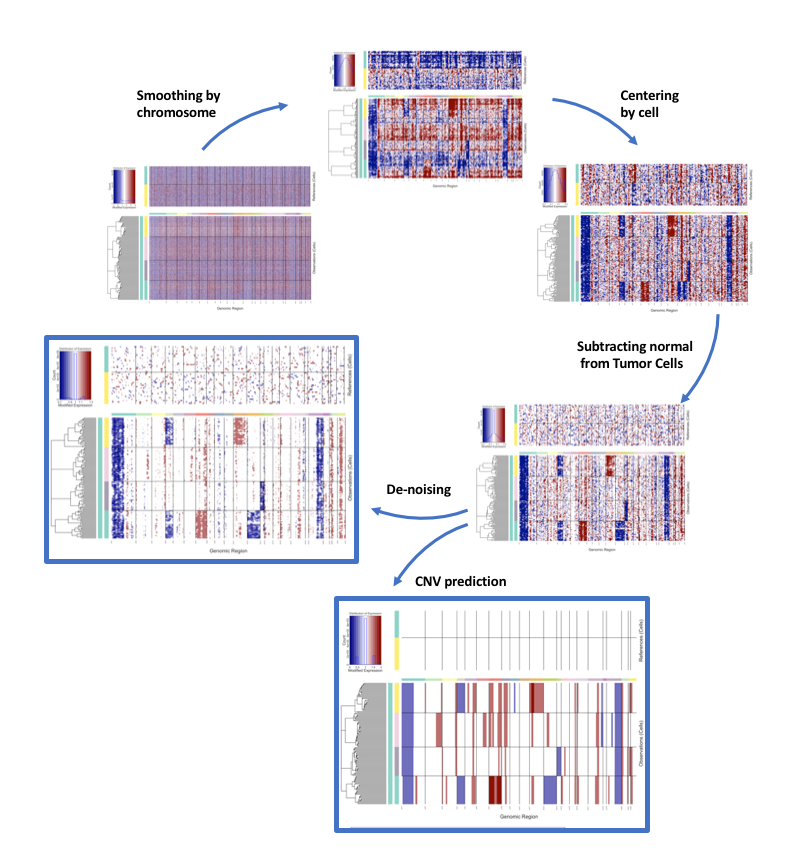
Setting run(denoise=TRUE) enables the de-noising procedure. Several de-noising filters are available for exploration.
Setting run(HMM=TRUE) enables the CNV predictions. There are multiple inferCNV HMM prediction methods available to explore as well.
算法实现
过滤基因:从计数矩阵中删除那些表达少于’min_cells_per_gene’的基因。min_cells_per_gene=3
测序深度的标准化
对数转换:将单个矩阵值(x)变换为log(x+1)
所有细胞中减去reference平均值。因为这个减法是在对数空间中执行的,所以这实际上减去的是log(fold_change)
对数倍变化值的动态范围阈值。 abs(log(x + 1))超过“ max_centered_threshold”(默认值= 3)的任何值均以该值为上限。
染色体级平滑:对于每一个细胞,沿着每条染色体排列的基因的表达强度通过加权平均进行平滑处理。默认情况下,这是一个包含101个基因的窗口,它们的权重是金字塔形的。
居中细胞:在大多数基因不在CNV区域的假设下,每个细胞的中值表达强度为零。
相对于正常细胞的调整:再次从肿瘤细胞中减去正常平均值。这进一步补偿了平滑处理后产生的差异。
对数变换被还原。
以上会生成“初步推断对象”。支持CNVs的信号通常在图中很明显。可以用de-noising提高信噪比。同样,可以使用HMM预测CNV区域。
资源消耗统计
6017个细胞,20900个基因。cpu=09:06:33, maxvmem=36.923G
其他
如果要修改heatmap的布局,需要修改.plot_observations_layout函数中obs_lmat、obs_lhei的值,obs_lmat是一个矩阵,它将画布分成了几个部分,obs_lhei用来调节行高。