[TOC]
标准流程

检查md5值
1 | md5sum -c checksums.md5 |
trimmomatic
1 | 2019-2-12 17:16:35 trim |
2019-2-13 12:55:38 查看结果,只trim了一部分,查看trim.log。HB-9-3终止。
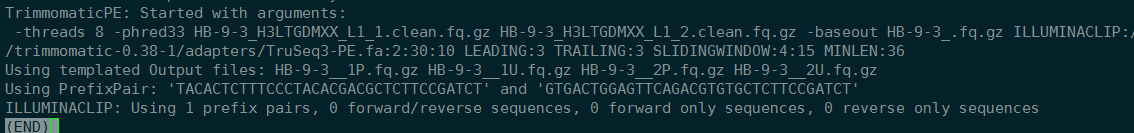
ps -aux | grep trimmomatic 查看任务是否还在运行。没有在运行。程序不知什么原因意外中断,来的时候xshell也没有连上服务器,怀疑是服务器自动重启了。
1 | nohup mv HB-*.fq.gz trim & |
2019-2-13 17:06:38 查看任务,意外终止。NH-11-2
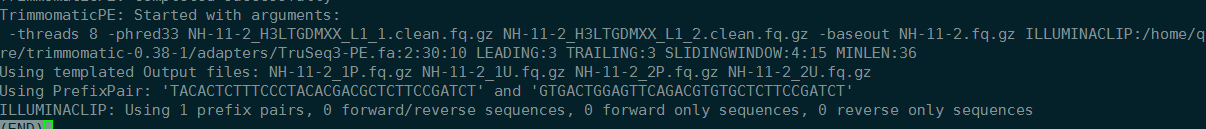
移动、删除文件,reboot,重新执行脚本。
1 | nohup time bash trim.sh >> trim.log 2>&1 & |
2019-2-13 19:35:51 查看,又意外停止
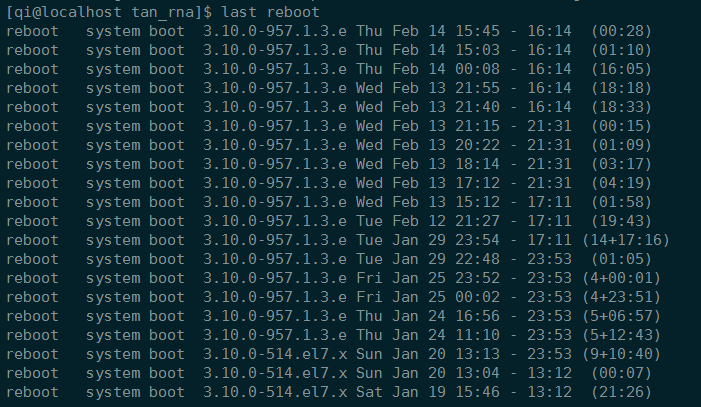
查看系统最后重启时间
1 | who -b |

显然,服务器会自动重启。
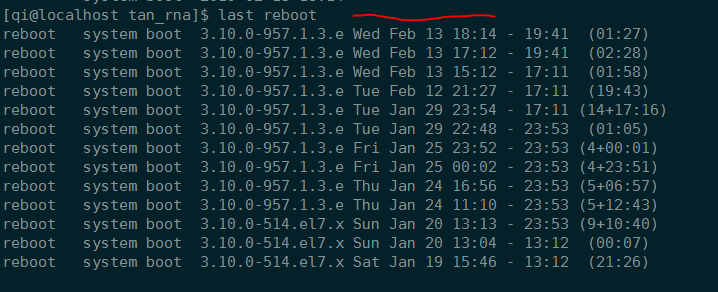
不运行任务的时候,不会重启。是因为CPU温度过高?
更改脚本试一试,把-threads 8 改成 4
1 | for i in *_1.clean.fq.gz |
1 | nohup time bash trim.sh > trim.log 2>&1 & |
没有效果
threads改成16
一小时不到又重启了。。
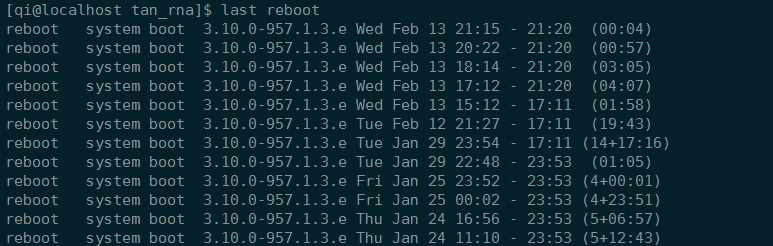
2019-2-14 16:16:57
质检
1 | fastqc *.fq.gz -o qcDIR/ -t 6 -d qcDIR/ >>fastqc.r.log 2>>fastqc.e.log |
1 | conda install -c bioconda multiqc |
做multiqc之前构建一个python3的虚拟环境
1 | python --version |
1 | 利用multiqc整合结果,方便批量查看 |
用 snakemake 编写任务流程
snakemake是一个用来编写任务流程的工具,用python写的,因此其执行的流程脚本也比较通俗易懂,易于理解,可以看做用户友好版的make。(make在安装软件的时候会用到,没有研究过makefile文件,对它的用处不是太了解。)
其实流程控制是复杂任务(在生信领域很常见)必需的关注点。只是snakemake对于代码功力不够的人来说,在写好代码与重复流程的花销的trade off上,还不如一遍遍重复流程。。但是真的是一个写好了就很好用的东西。
snakemake能够使用文件名通配的方式对一类文件进行处理
1 | installing |
简单的例子
1 | cd $HOME |
snakemake 脚本
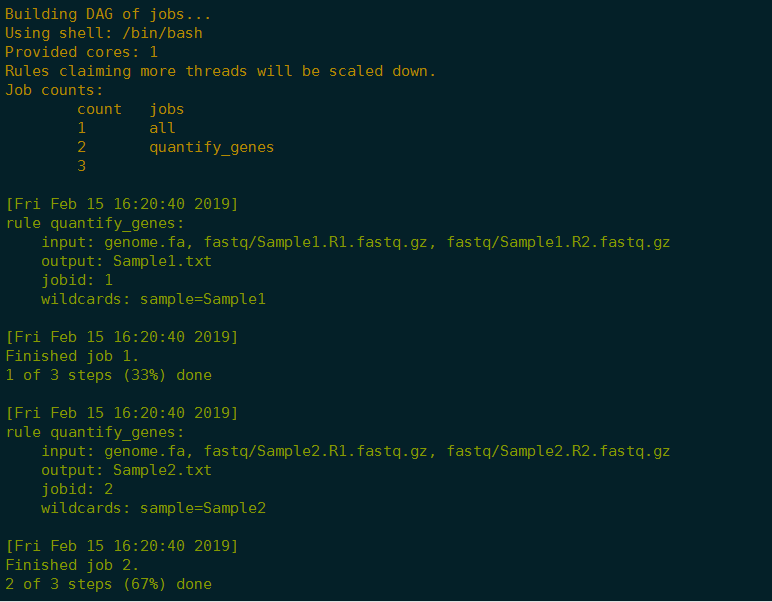
1 | SAMPLES = ['Sample1', 'Sample2'] |
学院集群上运行
1 | snakemake --snakefile Snakefile |
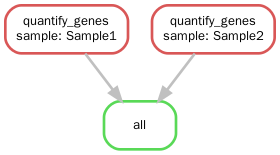
1 | 可视化 |
snakemake 规则
- Snakemake基于规则执行命令,规则一般由input, output,shell三部分组成。除了rule all,其余必须有output
- Snakemake可以自动确定不同规则的输入输出的依赖关系,根据时间戳来判断文件是否需要重新生成
- Snakemake以{sample}.fa形式进行文件名通配,用{wildcards.sample}获取sample的实际文件名
- Snakemake用expand()生成多个文件名,本质是Python的列表推导式
- Snakemake可以在规则外直接写Python代码,在规则内的run里也可以写Python代码。
- Snakefile的第一个规则通常是rule all,根据all里的文件决定执行哪些rule。如上面的例子,注释掉all里的input则不执行第二条rule,(推断未尝试:rule all里定义最终的输出文件,程序也能执行,那么rule里的输出文件在什么时候会被删除,是在所有rule运行完之后,还是在判断出该输出文件不会被用到的时候?)
- 在output中的结果文件可以是未存在目录中的文件,这时会自动创建不存在的目录(不需要事先建文件夹,这个功能实在是方便)
snakemake 命令
wildcards: 用来获取通配符匹配到的部分,例如对于通配符”{dataset}/file.{group}.txt”匹配到文件101/file.A.txt,则{wildcards.dataset}就是101,{wildcards.group}就是A。
temp: 通过temp方法可以在所有rule运行完后删除指定的中间文件,eg.output: temp(“f1.bam”)。
protected: 用来指定某些中间文件是需要保留的,eg.output: protected(“f1.bam”)。
snakemake 执行
一般讲所有的参数配置写入Snakefile后直接在Snakefile所在路径执行snakemake命令即可开始执行流程任务,如果只有一个snakefile的话,连文件都不用写。一些常用的参数:
1 | --snakefile, -s 指定Snakefile,否则是当前目录下的Snakefile |
snakemake 实践
1 | # 2019-2-19 10:28:52 part |
实验室服务器试运行
- By default snakemake executes the first rule in the snakefile.
- output中会自动创建没有的文件夹
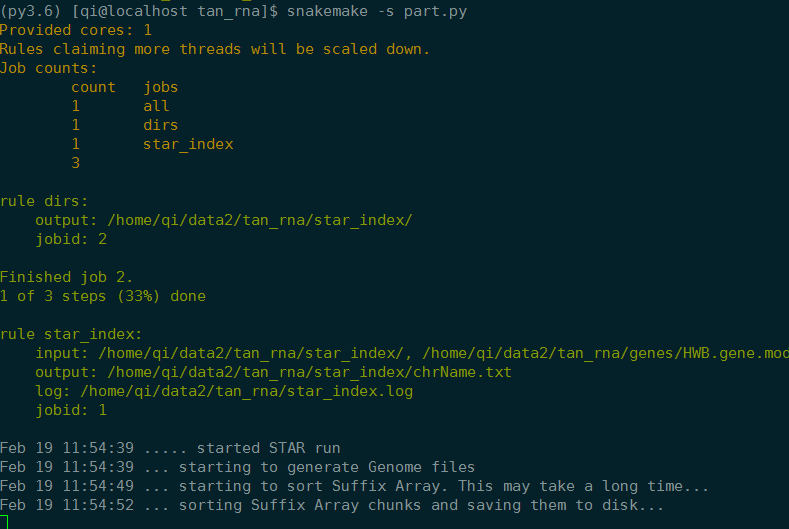
1 | cores设置核心数 |
跑samples中的所有样品
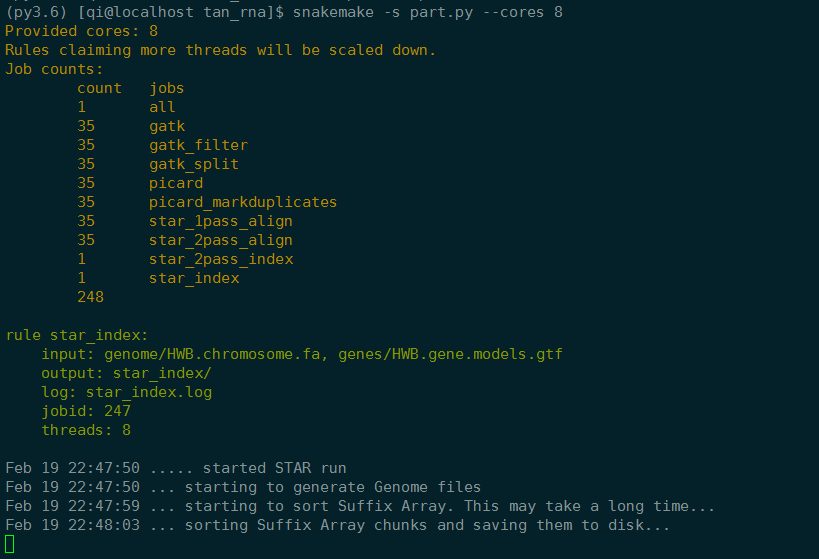
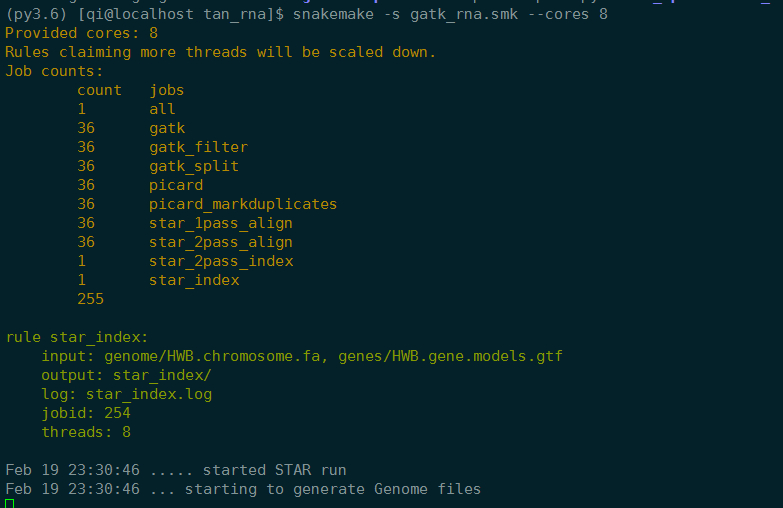
存储空间不够,停止
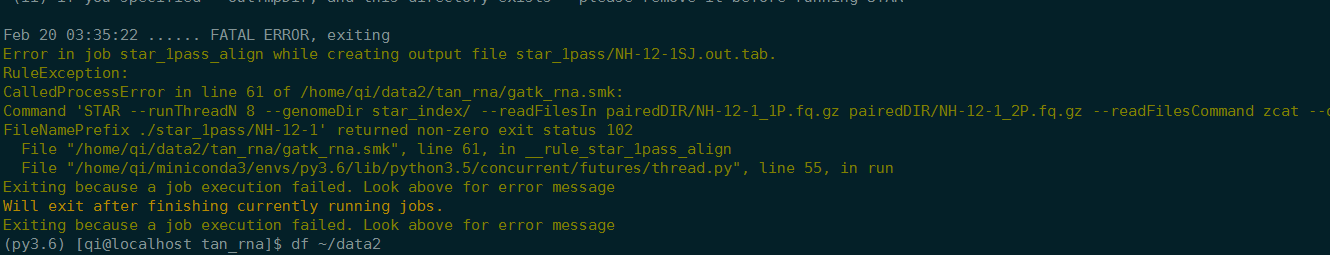
重新运行之后,是从NH-12-1开始
之前的结果只有NH-12-1Log.out,时间是2019/2/20 3:35 现在是:
说明这个样本从头开始跑了。
集群上运行
1 | 查看节点状态 |
1 | 查看错误信息 输出目录要用 directory() ,在实验室服务器上没有遇上这种情况 |
ImproperOutputException in line 30 of /home02/qizhengyang/qizhengyang/gatk_rna/Snakefile:
Outputs of incorrect type (directories when expecting files or vice versa). Output directories must be flagged with directory(). for rule star_index:
star_index/
Removing output files of failed job star_index since they might be corrupted:
star_index/
Skipped removing non-empty directory star_index/
Shutting down, this might take some time.
1 | snakemake -s Snakefile1 --cluster "qsub -V -q high" -j 20 |
2019-2-20 20:22:01 错误:
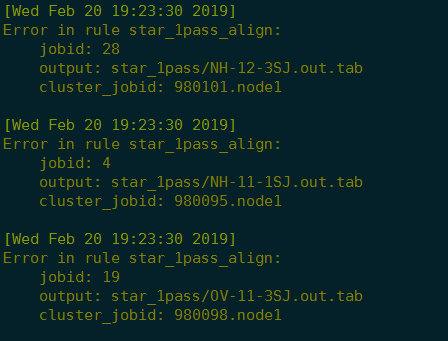
1 | less snakejob.star_1pass_align.28.sh.e980101 |
Possible cause 1: not enough RAM. Check if you have enough RAM 6975932175 bytes
1 | 剩下的三个重新跑 |
没有安装picard。。这种低级错误千万不能再犯了
1 | conda install -c bioconda picard |
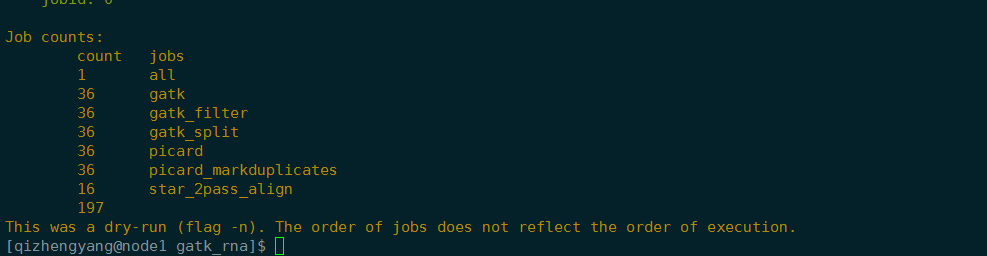
终端意外退出,输出到屏幕上的信息没有了
1 | ls -a |
1 | snakemake -s Snakefile1 --cluster "qsub -V -q high" -j 20 |
Error: Directory cannot be locked. Please make sure that no other Snakemake process is trying to create the same files in the following directory
1 | 解决办法 https://zhuanlan.zhihu.com/p/47575136 |
报错
1 | less snakejob.star_2pass_align.68.sh.e980306 |
libgcc_s.so.1 must be installed for pthread_cancel to work
1 | less snakejob.star_2pass_align.68.sh.e980306 |
短暂的断网,与服务器连接中断
1 | snakemake -n |
nohup将程序放入后台
1 | whatis nohup |
If standard input is a terminal, redirect it from /dev/null. If standard output is a terminal, append output to ‘nohup.out’ if possible, ‘$HOME/nohup.out’ otherwise. If standard error is a terminal, redirect it to standard output. To save output to FILE, use ‘nohup COMMAND > FILE’.
重定向与后台运行的知识
1 | nohup snakemake -n & |
1 | nohup方式后台运行(&) 忽略所有发送给子命令的挂断(SIGHUP)信号 重定向子命令的标准输出(stdout)和标准错误(stderr) |
IP 意外变化
2019-2-21 16:45:01 实验室的服务器连不上,我查看了ssh server是否开启,查了IP,发现是IP变了。
重新开始
2019-2-21 21:33:58
查看了实验室服务器生的Index文件夹,比较了大小,之前第一步需要重做。。很悲惨
gatk加了 -Xmx10g -jar # Decrease Java heap size (-Xmx/-Xms)
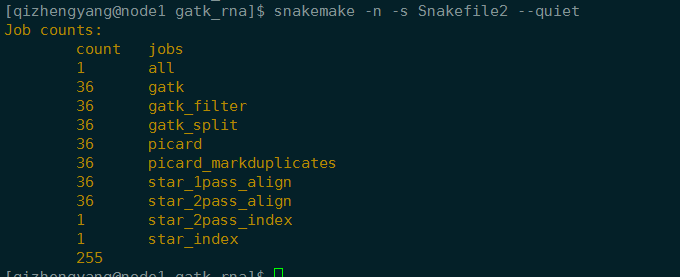
1 | snakemake -n -s Snakefile2 --quiet |
1 | snakemake --unlock |
之后再运行命令
1 | nohup snakemake -s Snakefile2 --cluster "qsub -V -q high" -j 10 &> snakemake.out & |
2019-2-22 14:56:03 查看任务
1 | ps aux|grep Snakefile2|grep -v grep |

这个任务在内存中,但 qstat没有任务在运行
1 | less snakemake.out |
这个进程kill不掉 kill 7550
但是,好像起作用了,log文件时间改了,似乎多了一句话(下次log文件应该copy一份下来,或者将最后的内容粘贴下来)
Will exit after finishing currently running jobs.
重新运行
1 | snakemake --unlock |
kill -9 7550 有效果
两个进程信息比较

院里集群上的时间快18分钟
报错先查看snakemake.log,然后查看job的std err
star 2pass align 出错
2019-2-22 20:06:58
1 | 查看错误信息 |
Building DAG of jobs…
Using shell: /bin/bash
Provided cores: 20
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 star_2pass_align
1[Fri Feb 22 16:44:54 2019]
rule star_2pass_align:
input: star_index_2pass/, pairedDIR/OV-9-2_1P.fq.gz, pairedDIR/OV-9-2_2P.fq.gz
output: star_2pass/OV-9-2Aligned.out.sam
jobid: 0
wildcards: sample=OV-9-2
threads: 20libgcc_s.so.1 must be installed for pthread_cancel to work
terminate called after throwing an instance of ‘St9bad_alloc’
libgcc_s.so.1 must be installed for pthread_cancel to work
what(): std::bad_alloc
[Fri Feb 22 16:46:26 2019]
Error in rule star_2pass_align:
jobid: 0
output: star_2pass/OV-9-2Aligned.out.samRuleException:
CalledProcessError in line 101 of /home02/qizhengyang/qizhengyang/gatk_rna/Snakefile2:
Command ‘set -euo pipefail; STAR –runThreadN 20 –genomeDir star_index_2pass/ –readFilesIn pairedDIR/OV-9-2_1P.fq.gz pairedDIR/OV-9-2_2P.fq.gz –readFilesCommand zcat –outFileNamePrefix star_2pass/OV-9-2’ returned non-zero exit status 141.
snakejob.star_2pass_align.52.sh.e980754
1 | 查看任务 |
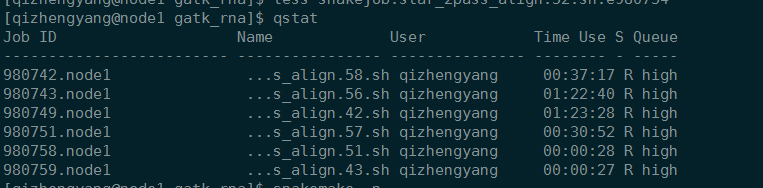
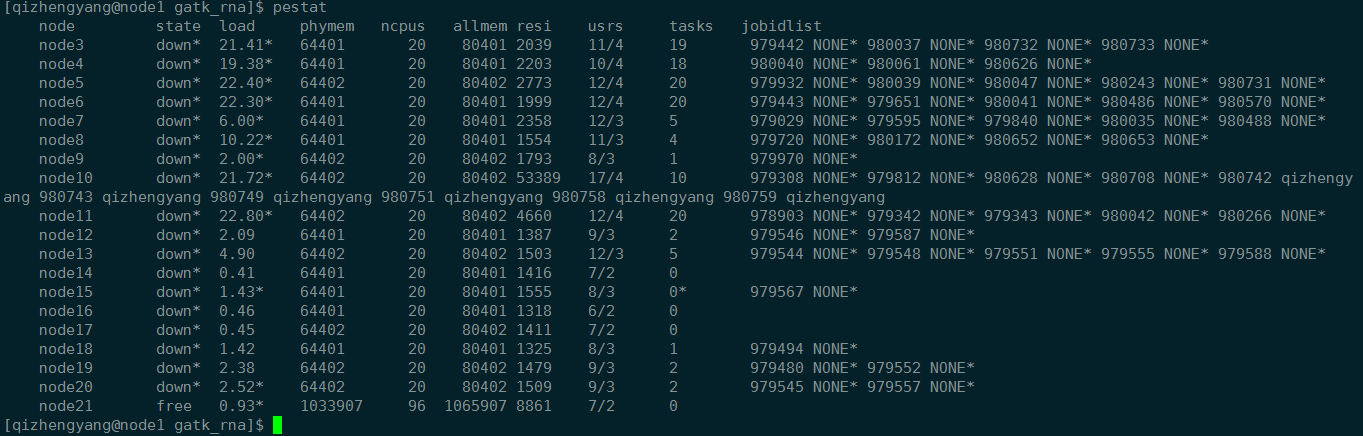
1 | pbsnodes -a | less |
[qizhengyang@node1 gatk_rna]$
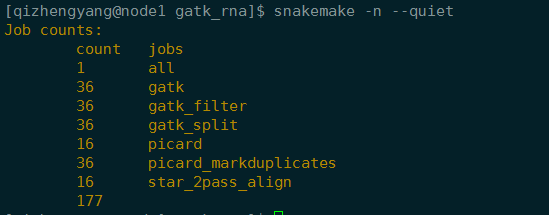
snakemake -n
Building DAG of jobs…
IncompleteFilesException:
The files below seem to be incomplete. If you are sure that certain files are not incomplete, mark them as complete withsnakemake --cleanup-metadata <filenames>To re-generate the files rerun your command with the –rerun-incomplete flag.
Incomplete files:
star_2pass/OV-10-1Aligned.out.sam
star_2pass/NH-10-3Aligned.out.sam
star_2pass/OV-10-2Aligned.out.sam
star_2pass/NH-11-2Aligned.out.sam
star_2pass/HB-9-3Aligned.out.sam
star_2pass/HB-10-3Aligned.out.sam
这样是在当前节点运行的
[qizhengyang@node1 gatk_rna]$
snakemake --rerun-incomplete
Building DAG of jobs…
Using shell: /bin/bash
Provided cores: 1
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 all
36 gatk
36 gatk_filter
36 gatk_split
36 picard
36 picard_markduplicates
25 star_2pass_align
206[Fri Feb 22 23:06:04 2019]
rule picard:
input: star_2pass/OV-12-3Aligned.out.sam
output: star_2pass/OV-12-3_rg_added_sorted.bam
jobid: 101
wildcards: sample=OV-12-3Job counts:
count jobs
1 picard
1
INFO 2019-02-22 23:06:07 AddOrReplaceReadGroups
1 | 只有胖节点有用,将任务提交到胖节点上,比需指定 -q low。默认是 high,会处于排队状态 |
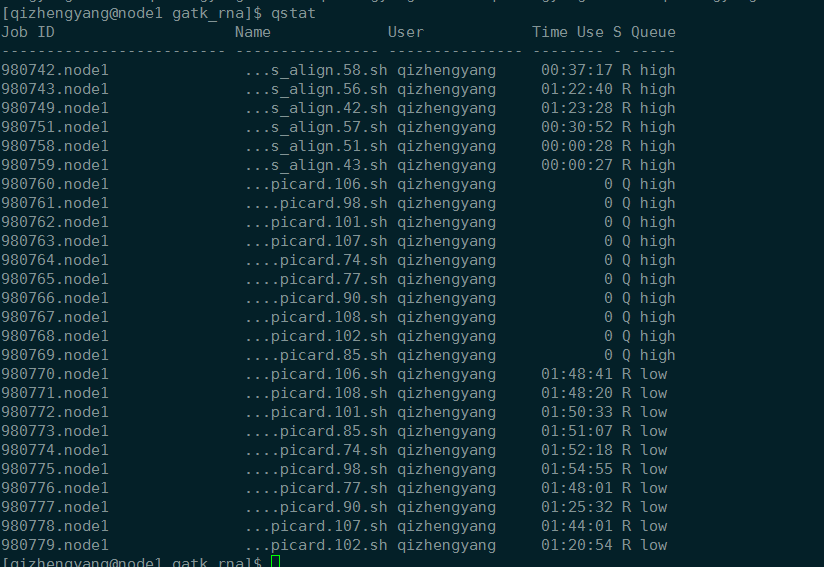
[qizhengyang@node1 gatk_rna]$ snakemake –rerun-incomplete –cluster “qsub -q low” –jobs 10
Building DAG of jobs…
Using shell: /bin/bash
Provided cluster nodes: 10
Job counts:
count jobs
1 all
36 gatk
36 gatk_filter
36 gatk_split
36 picard
36 picard_markduplicates
25 star_2pass_align
206[Fri Feb 22 23:24:56 2019]
rule picard:
input: star_2pass/HB-12-1Aligned.out.sam
output: star_2pass/HB-12-1_rg_added_sorted.bam
jobid: 106
wildcards: sample=HB-12-1Submitted job 106 with external jobid ‘980770.node1’.
[Fri Feb 22 23:24:56 2019]
rule picard:
input: star_2pass/NH-9-2Aligned.out.sam
output: star_2pass/NH-9-2_rg_added_sorted.bam
jobid: 108
wildcards: sample=NH-9-2Submitted job 108 with external jobid ‘980771.node1’.
[Fri Feb 22 23:24:56 2019]
rule picard:
input: star_2pass/OV-12-3Aligned.out.sam
output: star_2pass/OV-12-3_rg_added_sorted.bam
jobid: 101
wildcards: sample=OV-12-3Submitted job 101 with external jobid ‘980772.node1’.
[Fri Feb 22 23:24:56 2019]
rule picard:
input: star_2pass/HB-10-1Aligned.out.sam
output: star_2pass/HB-10-1_rg_added_sorted.bam
jobid: 85
wildcards: sample=HB-10-1Submitted job 85 with external jobid ‘980773.node1’.
[Fri Feb 22 23:24:56 2019]
rule picard:
input: star_2pass/NH-9-3Aligned.out.sam
output: star_2pass/NH-9-3_rg_added_sorted.bam
jobid: 74
wildcards: sample=NH-9-3Submitted job 74 with external jobid ‘980774.node1’.
[Fri Feb 22 23:24:56 2019]
rule picard:
input: star_2pass/OV-10-3Aligned.out.sam
output: star_2pass/OV-10-3_rg_added_sorted.bam
jobid: 98
wildcards: sample=OV-10-3Submitted job 98 with external jobid ‘980775.node1’.
[Fri Feb 22 23:24:57 2019]
rule picard:
input: star_2pass/HB-11-2Aligned.out.sam
output: star_2pass/HB-11-2_rg_added_sorted.bam
jobid: 77
wildcards: sample=HB-11-2Submitted job 77 with external jobid ‘980776.node1’.
[Fri Feb 22 23:24:57 2019]
rule picard:
input: star_2pass/OV-11-2Aligned.out.sam
output: star_2pass/OV-11-2_rg_added_sorted.bam
jobid: 90
wildcards: sample=OV-11-2Submitted job 90 with external jobid ‘980777.node1’.
[Fri Feb 22 23:24:57 2019]
rule picard:
input: star_2pass/NH-12-1Aligned.out.sam
output: star_2pass/NH-12-1_rg_added_sorted.bam
jobid: 107
wildcards: sample=NH-12-1Submitted job 107 with external jobid ‘980778.node1’.
[Fri Feb 22 23:24:57 2019]
rule picard:
input: star_2pass/OV-12-1Aligned.out.sam
output: star_2pass/OV-12-1_rg_added_sorted.bam
jobid: 102
wildcards: sample=OV-12-1Submitted job 102 with external jobid ‘980779.node1’.
运行时间
picard 半小时,CPU时间是4小时。但是picard我并没有设置使用多少线程。
qstat
qstat -a
qstat -n
2019-2-23 11:02:59
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
Complete log: /home02/qizhengyang/qizhengyang/gatk_rna/.snakemake/log/2019-02-22T232454.376651.snakemake.log
出错的任务
[Sat Feb 23 01:21:32 2019]
Error in rule picard:
jobid: 88
output: star_2pass/OV-9-2_rg_added_sorted.bam
cluster_jobid: 980797.node1
1 | less snakejob.picard.88.sh.e980797 |
Building DAG of jobs…
Using shell: /bin/bash
Provided cores: 96
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 picard
1[Sat Feb 23 00:58:20 2019]
rule picard:
input: star_2pass/OV-9-2Aligned.out.sam
output: star_2pass/OV-9-2_rg_added_sorted.bam
jobid: 0
wildcards: sample=OV-9-2INFO 2019-02-23 00:58:23 AddOrReplaceReadGroups
** NOTE: Picard’s command line syntax is changing.
** For more information, please see:
** https://github.com/broadinstitute/picard/wiki/Command-Line-Syntax-Transition-For-Users-(Pre-Transition)
** The command line looks like this in the new syntax:
** AddOrReplaceReadGroups -I star_2pass/OV-9-2Aligned.out.sam -O star_2pass/OV-9-2_rg_added_sorted.bam -SO coordinate -RGID OV-9-2 -RGLB rna -RGPL illumina -RGPU hiseq -RGSM OV-9-2
00:58:23.874 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/home02/qizhengyang/anaconda3/share/picard-2.18.26-0/picard.jar!/
com/intel/gkl/native/libgkl_compression.so
[Sat Feb 23 00:58:23 CST 2019] AddOrReplaceReadGroups INPUT=star_2pass/OV-9-2Aligned.out.sam OUTPUT=star_2pass/OV-9-2_rg_added_sorted.bam SORT_ORDER=c
oordinate RGID=OV-9-2 RGLB=rna RGPL=illumina RGPU=hiseq RGSM=OV-9-2 VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX
_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json USE_JDK_DEFLATER=false USE_JDK_INFLATER=false
[Sat Feb 23 00:58:23 CST 2019] Executing as qizhengyang@node21 on Linux 2.6.32-431.el6.x86_64 amd64; OpenJDK 64-Bit Server VM 1.8.0_121-b15; Deflater:
Intel; Inflater: Intel; Provider GCS is not available; Picard version: 2.18.26-SNAPSHOT
INFO 2019-02-23 00:58:27 AddOrReplaceReadGroups Created read-group ID=OV-9-2 PL=illumina LB=rna SM=OV-9-2INFO 2019-02-23 00:59:00 AddOrReplaceReadGroups Processed 1,000,000 records. Elapsed time: 00:00:33s. Time for last 1,000,000: 33s. L
ast read position: chr9:26,425,698
INFO 2019-02-23 00:59:34 AddOrReplaceReadGroups Processed 2,000,000 records. Elapsed time: 00:01:07s. Time for last 1,000,000: 33s. L
ast read position: chr5:38,200,263
INFO 2019-02-23 01:00:29 AddOrReplaceReadGroups Processed 3,000,000 records. Elapsed time: 00:02:02s. Time for last 1,000,000: 55s. L
ast read position: chr4:1,886,332
INFO 2019-02-23 01:00:56 AddOrReplaceReadGroups Processed 4,000,000 records. Elapsed time: 00:02:28s. Time for last 1,000,000: 26s. L
ast read position: chr5:6,379,073
INFO 2019-02-23 01:01:34 AddOrReplaceReadGroups Processed 5,000,000 records. Elapsed time: 00:03:07s. Time for last 1,000,000: 38s. L
ast read position: chr6:18,567,116
INFO 2019-02-23 01:02:13 AddOrReplaceReadGroups Processed 6,000,000 records. Elapsed time: 00:03:46s. Time for last 1,000,000: 38s. L
ast read position: chr9:39,573,973
INFO 2019-02-23 01:02:40 AddOrReplaceReadGroups Processed 7,000,000 records. Elapsed time: 00:04:13s. Time for last 1,000,000: 27s. L
ast read position: chr5:22,938,417
INFO 2019-02-23 01:03:19 AddOrReplaceReadGroups Processed 8,000,000 records. Elapsed time: 00:04:51s. Time for last 1,000,000: 38s. L
ast read position: chr4:23,545,960
INFO 2019-02-23 01:03:55 AddOrReplaceReadGroups Processed 9,000,000 records. Elapsed time: 00:05:28s. Time for last 1,000,000: 36s. L
ast read position: chr4:17,586,047
INFO 2019-02-23 01:04:24 AddOrReplaceReadGroups Processed 10,000,000 records. Elapsed time: 00:05:57s. Time for last 1,000,000: 28s. L
ast read position: chr7:21,260,263
INFO 2019-02-23 01:04:58 AddOrReplaceReadGroups Processed 11,000,000 records. Elapsed time: 00:06:31s. Time for last 1,000,000: 33s. L
ast read position: chr4:25,569,847
INFO 2019-02-23 01:05:36 AddOrReplaceReadGroups Processed 12,000,000 records. Elapsed time: 00:07:08s. Time for last 1,000,000: 37s. L
ast read position: chr4:25,075,124
INFO 2019-02-23 01:06:04 AddOrReplaceReadGroups Processed 13,000,000 records. Elapsed time: 00:07:36s. Time for last 1,000,000: 28s. L
ast read position: chr1:5,055,820
INFO 2019-02-23 01:06:38 AddOrReplaceReadGroups Processed 14,000,000 records. Elapsed time: 00:08:11s. Time for last 1,000,000: 34s. L
ast read position: chr5:40,818,516
INFO 2019-02-23 01:07:13 AddOrReplaceReadGroups Processed 15,000,000 records. Elapsed time: 00:08:46s. Time for last 1,000,000: 34s. L
ast read position: chr3:28,597,426
INFO 2019-02-23 01:07:40 AddOrReplaceReadGroups Processed 16,000,000 records. Elapsed time: 00:09:12s. Time for last 1,000,000: 26s. L
ast read position: chr2:52,066,083
INFO 2019-02-23 01:08:24 AddOrReplaceReadGroups Processed 17,000,000 records. Elapsed time: 00:09:54s. Time for last 1,000,000: 41s. L
ast read position: chr5:8,214,376
INFO 2019-02-23 01:08:53 AddOrReplaceReadGroups Processed 18,000,000 records. Elapsed time: 00:10:25s. Time for last 1,000,000: 31s. L
ast read position: chr2:42,040,926
INFO 2019-02-23 01:09:19 AddOrReplaceReadGroups Processed 19,000,000 records. Elapsed time: 00:10:52s. Time for last 1,000,000: 26s. L
ast read position: chr8:3,160,150
INFO 2019-02-23 01:09:59 AddOrReplaceReadGroups Processed 20,000,000 records. Elapsed time: 00:11:32s. Time for last 1,000,000: 39s. L
ast read position: chr9:2,765,258
INFO 2019-02-23 01:10:36 AddOrReplaceReadGroups Processed 21,000,000 records. Elapsed time: 00:12:08s. Time for last 1,000,000: 36s. L
ast read position: chr7:872,502
INFO 2019-02-23 01:11:13 AddOrReplaceReadGroups Processed 22,000,000 records. Elapsed time: 00:12:46s. Time for last 1,000,000: 37s. L
ast read position: chr5:38,922,782
INFO 2019-02-23 01:11:43 AddOrReplaceReadGroups Processed 23,000,000 records. Elapsed time: 00:13:15s. Time for last 1,000,000: 29s. L
ast read position: chr2:43,156,703
INFO 2019-02-23 01:12:10 AddOrReplaceReadGroups Processed 24,000,000 records. Elapsed time: 00:13:42s. Time for last 1,000,000: 26s. L
ast read position: chr2:42,168,429
INFO 2019-02-23 01:12:37 AddOrReplaceReadGroups Processed 25,000,000 records. Elapsed time: 00:14:10s. Time for last 1,000,000: 27s. L
ast read position: chr5:47,894,574
INFO 2019-02-23 01:13:05 AddOrReplaceReadGroups Processed 26,000,000 records. Elapsed time: 00:14:37s. Time for last 1,000,000: 27s. L
ast read position: chr5:47,836,828
INFO 2019-02-23 01:13:33 AddOrReplaceReadGroups Processed 27,000,000 records. Elapsed time: 00:15:05s. Time for last 1,000,000: 27s. L
ast read position: chr8:19,290,720
INFO 2019-02-23 01:14:01 AddOrReplaceReadGroups Processed 28,000,000 records. Elapsed time: 00:15:33s. Time for last 1,000,000: 27s. L
ast read position: chr5:17,045,282
INFO 2019-02-23 01:14:29 AddOrReplaceReadGroups Processed 29,000,000 records. Elapsed time: 00:16:02s. Time for last 1,000,000: 28s. L
ast read position: chr4:16,374,034
INFO 2019-02-23 01:14:56 AddOrReplaceReadGroups Processed 30,000,000 records. Elapsed time: 00:16:28s. Time for last 1,000,000: 26s. L
ast read position: chr2:51,915,818
INFO 2019-02-23 01:15:24 AddOrReplaceReadGroups Processed 31,000,000 records. Elapsed time: 00:16:57s. Time for last 1,000,000: 28s. L
ast read position: chr3:24,910,383
INFO 2019-02-23 01:15:55 AddOrReplaceReadGroups Processed 32,000,000 records. Elapsed time: 00:17:27s. Time for last 1,000,000: 30s. L
ast read position: chr7:12,459,410
INFO 2019-02-23 01:16:22 AddOrReplaceReadGroups Processed 33,000,000 records. Elapsed time: 00:17:54s. Time for last 1,000,000: 26s. L
ast read position: chr9:39,689,303
[Sat Feb 23 01:20:04 CST 2019] picard.sam.AddOrReplaceReadGroups done. Elapsed time: 21.69 minutes.
Runtime.totalMemory()=1004011520
To get help, see http://broadinstitute.github.io/picard/index.html#GettingHelp
Exception in thread “main” java.lang.OutOfMemoryError: GC overhead limit exceeded
at java.util.Arrays.copyOfRange(Arrays.java:3664)
at java.lang.String.(String.java:207)
at java.lang.String.substring(String.java:1969)
at htsjdk.samtools.util.StringUtil.split(StringUtil.java:89)
at htsjdk.samtools.SAMLineParser.parseLine(SAMLineParser.java:229)
at htsjdk.samtools.SAMTextReader$RecordIterator.parseLine(SAMTextReader.java:268)
at htsjdk.samtools.SAMTextReader$RecordIterator.next(SAMTextReader.java:255)
at htsjdk.samtools.SAMTextReader$RecordIterator.next(SAMTextReader.java:228)
at htsjdk.samtools.SamReader$AssertingIterator.next(SamReader.java:569)
at htsjdk.samtools.SamReader$AssertingIterator.next(SamReader.java:548)
at picard.sam.AddOrReplaceReadGroups.doWork(AddOrReplaceReadGroups.java:182)
at picard.cmdline.CommandLineProgram.instanceMain(CommandLineProgram.java:295)
at picard.cmdline.PicardCommandLine.instanceMain(PicardCommandLine.java:103)
at picard.cmdline.PicardCommandLine.main(PicardCommandLine.java:113)
1 picard
1[Sat Feb 23 00:58:20 2019]
rule picard:
input: star_2pass/OV-9-2Aligned.out.sam
output: star_2pass/OV-9-2_rg_added_sorted.bam
jobid: 0
wildcards: sample=OV-9-2INFO 2019-02-23 00:58:23 AddOrReplaceReadGroups
** NOTE: Picard’s command line syntax is changing.
** For more information, please see:
** https://github.com/broadinstitute/picard/wiki/Command-Line-Syntax-Transition-For-Users-(Pre-Transition)
** The command line looks like this in the new syntax:
** AddOrReplaceReadGroups -I star_2pass/OV-9-2Aligned.out.sam -O star_2pass/OV-9-2_rg_added_sorted.bam -SO coordinate -RGID OV-9-2 -RGLB rn
a -RGPL illumina -RGPU hiseq -RGSM OV-9-2
00:58:23.874 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/home02/qizhengyang/anaconda3/share/picard-2.18.26-0/picard.jar!/
com/intel/gkl/native/libgkl_compression.so
[Sat Feb 23 00:58:23 CST 2019] AddOrReplaceReadGroups INPUT=star_2pass/OV-9-2Aligned.out.sam OUTPUT=star_2pass/OV-9-2_rg_added_sorted.bam SORT_ORDER=c
oordinate RGID=OV-9-2 RGLB=rna RGPL=illumina RGPU=hiseq RGSM=OV-9-2 VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX
_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json USE_JDK_DEFLATER=false USE_JDK_INFLATER=false
[Sat Feb 23 00:58:23 CST 2019] Executing as qizhengyang@node21 on Linux 2.6.32-431.el6.x86_64 amd64; OpenJDK 64-Bit Server VM 1.8.0_121-b15; Deflater:
Intel; Inflater: Intel; Provider GCS is not available; Picard version: 2.18.26-SNAPSHOT
INFO 2019-02-23 00:58:27 AddOrReplaceReadGroups Created read-group ID=OV-9-2 PL=illumina LB=rna SM=OV-9-2INFO 2019-02-23 00:59:00 AddOrReplaceReadGroups Processed 1,000,000 records. Elapsed time: 00:00:33s. Time for last 1,000,000: 33s. L
ast read position: chr9:26,425,698
INFO 2019-02-23 00:59:34 AddOrReplaceReadGroups Processed 2,000,000 records. Elapsed time: 00:01:07s. Time for last 1,000,000: 33s. L
ast read position: chr5:38,200,263
INFO 2019-02-23 01:00:29 AddOrReplaceReadGroups Processed 3,000,000 records. Elapsed time: 00:02:02s. Time for last 1,000,000: 55s. L
ast read position: chr4:1,886,332
INFO 2019-02-23 01:00:56 AddOrReplaceReadGroups Processed 4,000,000 records. Elapsed time: 00:02:28s. Time for last 1,000,000: 26s. L
ast read position: chr5:6,379,073
INFO 2019-02-23 01:01:34 AddOrReplaceReadGroups Processed 5,000,000 records. Elapsed time: 00:03:07s. Time for last 1,000,000: 38s. L
ast read position: chr6:18,567,116
INFO 2019-02-23 01:02:13 AddOrReplaceReadGroups Processed 6,000,000 records. Elapsed time: 00:03:46s. Time for last 1,000,000: 38s. L
ast read position: chr9:39,573,973
INFO 2019-02-23 01:02:40 AddOrReplaceReadGroups Processed 7,000,000 records. Elapsed time: 00:04:13s. Time for last 1,000,000: 27s. L
ast read position: chr5:22,938,417
INFO 2019-02-23 01:03:19 AddOrReplaceReadGroups Processed 8,000,000 records. Elapsed time: 00:04:51s. Time for last 1,000,000: 38s. L
ast read position: chr4:23,545,960
INFO 2019-02-23 01:03:55 AddOrReplaceReadGroups Processed 9,000,000 records. Elapsed time: 00:05:28s. Time for last 1,000,000: 36s. L
ast read position: chr4:17,586,047
INFO 2019-02-23 01:04:24 AddOrReplaceReadGroups Processed 10,000,000 records. Elapsed time: 00:05:57s. Time for last 1,000,000: 28s. L
ast read position: chr7:21,260,263
INFO 2019-02-23 01:04:58 AddOrReplaceReadGroups Processed 11,000,000 records. Elapsed time: 00:06:31s. Time for last 1,000,000: 33s. L
ast read position: chr4:25,569,847
INFO 2019-02-23 01:05:36 AddOrReplaceReadGroups Processed 12,000,000 records. Elapsed time: 00:07:08s. Time for last 1,000,000: 37s. L
ast read position: chr4:25,075,124
INFO 2019-02-23 01:06:04 AddOrReplaceReadGroups Processed 13,000,000 records. Elapsed time: 00:07:36s. Time for last 1,000,000: 28s. L
ast read position: chr1:5,055,820
INFO 2019-02-23 01:06:38 AddOrReplaceReadGroups Processed 14,000,000 records. Elapsed time: 00:08:11s. Time for last 1,000,000: 34s. L
ast read position: chr5:40,818,516
INFO 2019-02-23 01:07:13 AddOrReplaceReadGroups Processed 15,000,000 records. Elapsed time: 00:08:46s. Time for last 1,000,000: 34s. L
ast read position: chr3:28,597,426
INFO 2019-02-23 01:07:40 AddOrReplaceReadGroups Processed 16,000,000 records. Elapsed time: 00:09:12s. Time for last 1,000,000: 26s. L
ast read position: chr2:52,066,083
INFO 2019-02-23 01:08:24 AddOrReplaceReadGroups Processed 17,000,000 records. Elapsed time: 00:09:54s. Time for last 1,000,000: 41s. L
ast read position: chr5:8,214,376
INFO 2019-02-23 01:08:53 AddOrReplaceReadGroups Processed 18,000,000 records. Elapsed time: 00:10:25s. Time for last 1,000,000: 31s. L
ast read position: chr2:42,040,926
INFO 2019-02-23 01:09:19 AddOrReplaceReadGroups Processed 19,000,000 records. Elapsed time: 00:10:52s. Time for last 1,000,000: 26s. L
ast read position: chr8:3,160,150
INFO 2019-02-23 01:09:59 AddOrReplaceReadGroups Processed 20,000,000 records. Elapsed time: 00:11:32s. Time for last 1,000,000: 39s. L
ast read position: chr9:2,765,258
INFO 2019-02-23 01:10:36 AddOrReplaceReadGroups Processed 21,000,000 records. Elapsed time: 00:12:08s. Time for last 1,000,000: 36s. L
ast read position: chr7:872,502
INFO 2019-02-23 01:11:13 AddOrReplaceReadGroups Processed 22,000,000 records. Elapsed time: 00:12:46s. Time for last 1,000,000: 37s. L
ast read position: chr5:38,922,782
INFO 2019-02-23 01:11:43 AddOrReplaceReadGroups Processed 23,000,000 records. Elapsed time: 00:13:15s. Time for last 1,000,000: 29s. L
ast read position: chr2:43,156,703
INFO 2019-02-23 01:12:10 AddOrReplaceReadGroups Processed 24,000,000 records. Elapsed time: 00:13:42s. Time for last 1,000,000: 26s. L
ast read position: chr2:42,168,429
INFO 2019-02-23 01:12:37 AddOrReplaceReadGroups Processed 25,000,000 records. Elapsed time: 00:14:10s. Time for last 1,000,000: 27s. L
ast read position: chr5:47,894,574
INFO 2019-02-23 01:13:05 AddOrReplaceReadGroups Processed 26,000,000 records. Elapsed time: 00:14:37s. Time for last 1,000,000: 27s. L
ast read position: chr5:47,836,828
INFO 2019-02-23 01:13:33 AddOrReplaceReadGroups Processed 27,000,000 records. Elapsed time: 00:15:05s. Time for last 1,000,000: 27s. L
ast read position: chr8:19,290,720
INFO 2019-02-23 01:14:01 AddOrReplaceReadGroups Processed 28,000,000 records. Elapsed time: 00:15:33s. Time for last 1,000,000: 27s. L
ast read position: chr5:17,045,282
INFO 2019-02-23 01:14:29 AddOrReplaceReadGroups Processed 29,000,000 records. Elapsed time: 00:16:02s. Time for last 1,000,000: 28s. L
ast read position: chr4:16,374,034
INFO 2019-02-23 01:14:56 AddOrReplaceReadGroups Processed 30,000,000 records. Elapsed time: 00:16:28s. Time for last 1,000,000: 26s. L
ast read position: chr2:51,915,818
INFO 2019-02-23 01:15:24 AddOrReplaceReadGroups Processed 31,000,000 records. Elapsed time: 00:16:57s. Time for last 1,000,000: 28s. L
ast read position: chr3:24,910,383
INFO 2019-02-23 01:15:55 AddOrReplaceReadGroups Processed 32,000,000 records. Elapsed time: 00:17:27s. Time for last 1,000,000: 30s. L
ast read position: chr7:12,459,410
INFO 2019-02-23 01:16:22 AddOrReplaceReadGroups Processed 33,000,000 records. Elapsed time: 00:17:54s. Time for last 1,000,000: 26s. L
ast read position: chr9:39,689,303
[Sat Feb 23 01:20:04 CST 2019] picard.sam.AddOrReplaceReadGroups done. Elapsed time: 21.69 minutes.
Runtime.totalMemory()=1004011520
To get help, see http://broadinstitute.github.io/picard/index.html#GettingHelp
Exception in thread “main” java.lang.OutOfMemoryError: GC overhead limit exceeded
at java.util.Arrays.copyOfRange(Arrays.java:3664)
at java.lang.String.(String.java:207)
at java.lang.String.substring(String.java:1969)
at htsjdk.samtools.util.StringUtil.split(StringUtil.java:89)
at htsjdk.samtools.SAMLineParser.parseLine(SAMLineParser.java:229)
at htsjdk.samtools.SAMTextReader$RecordIterator.parseLine(SAMTextReader.java:268)
at htsjdk.samtools.SAMTextReader$RecordIterator.next(SAMTextReader.java:255)
at htsjdk.samtools.SAMTextReader$RecordIterator.next(SAMTextReader.java:228)
at htsjdk.samtools.SamReader$AssertingIterator.next(SamReader.java:569)
at htsjdk.samtools.SamReader$AssertingIterator.next(SamReader.java:548)
at picard.sam.AddOrReplaceReadGroups.doWork(AddOrReplaceReadGroups.java:182)
at picard.cmdline.CommandLineProgram.instanceMain(CommandLineProgram.java:295)
at picard.cmdline.PicardCommandLine.instanceMain(PicardCommandLine.java:103)
at picard.cmdline.PicardCommandLine.main(PicardCommandLine.java:113)
Exception in thread “Thread-0” java.lang.OutOfMemoryError: GC overhead limit exceeded
at java.util.Hashtable.(Hashtable.java:190)
at java.util.Hashtable.(Hashtable.java:211)
at java.util.Properties.(Properties.java:148)
at java.util.Properties.(Properties.java:140)
at java.util.logging.LogManager.reset(LogManager.java:1321)
at java.util.logging.LogManager$Cleaner.run(LogManager.java:239)
Exception in thread “Thread-1” java.lang.OutOfMemoryError: GC overhead limit exceeded
at sun.nio.fs.UnixFileAttributes.get(UnixFileAttributes.java:68)
at sun.nio.fs.UnixFileSystemProvider.implDelete(UnixFileSystemProvider.java:227)
at sun.nio.fs.AbstractFileSystemProvider.delete(AbstractFileSystemProvider.java:103)
at java.nio.file.Files.delete(Files.java:1126)
at htsjdk.samtools.util.nio.DeleteOnExitPathHook.runHooks(DeleteOnExitPathHook.java:57)
at htsjdk.samtools.util.nio.DeleteOnExitPathHook$$Lambda$34/780934299.run(Unknown Source)
at java.lang.Thread.run(Thread.java:745)
[Sat Feb 23 01:20:54 2019]
Error in rule picard:
jobid: 0
output: star_2pass/OV-9-2_rg_added_sorted.bamRuleException:
CalledProcessError in line 115 of /home02/qizhengyang/qizhengyang/gatk_rna/Snakefile:
Command ‘set -euo pipefail; picard AddOrReplaceReadGroups I=star_2pass/OV-9-2Aligned.out.sam O=star_2pass/OV-9-2_rg_added_sorted.bam SO=coordinate RG
ID=OV-9-2 RGLB=rna RGPL=illumina RGPU=hiseq RGSM=OV-9-2’ returned non-zero exit status 1.
File “/home02/qizhengyang/qizhengyang/gatk_rna/Snakefile”, line 115, in __rule_picard
File “/home02/qizhengyang/anaconda3/lib/python3.6/concurrent/futures/thread.py”, line 56, in run
Removing output files of failed job picard since they might be corrupted:
star_2pass/OV-9-2_rg_added_sorted.bam
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
(END)
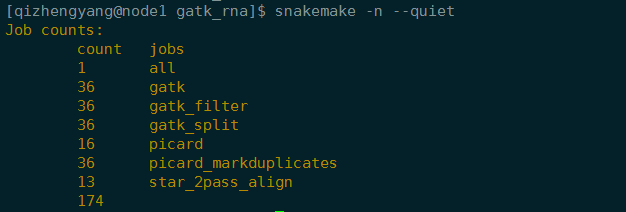
1 | snakemake -n --quiet |
一个终端与服务器连接终端
Socket error Event: 32 Error: 10053.
Connection closing…Socket close.Connection closed by foreign host.
Disconnected from remote host(qizhengyang) at 11:29:56.
Type `help’ to learn how to use Xshell prompt.
[C:~]$
last 显示用户最近登录信息。top用于实时显示 process 的动态。
1 | snakemake -n -s Snakefile2 --quiet |
[qizhengyang@node1 gatk_rna]$ jobs
[1]+ Running nohup snakemake –cluster “qsub -q low” –jobs 100 &
[qizhengyang@node1 gatk_rna]$ ps aux | grep snakemake | grep -v grep
528 39349 0.5 0.0 289360 26440 pts/5 Sl 14:00 0:01 /home02/qizhengyang/anaconda3/bin/python3.6 /home02/qizhengyang/anaconda3/bin/snakemake –cluster qsub -q low –jobs 100
1 | ll -h nohup.out |
一个 picard job 出现错误 Exception in thread “main” java.lang.OutOfMemoryError: GC overhead limit exceeded
Moreover, now i am running command like /usr/bin/java -Xmx40g -jar /usr/local/picard-tools-1.129/picard.jar …., and it’s working. Regards, Ravi.
ava刚刚出现的年代,有一个相比于其他语言的优势就是,内存回收机制。不需要明确的调用释放内存的API,java就自动完成,这个过程就是Garbage Collection,简称GC。
其实还是有一个终极方法的,而且是治标治本的方法,就是找到占用内存大的地方,把代码优化了,就不会出现这个问题了。
Building DAG of jobs…
Using shell: /bin/bash
Provided cores: 96
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 picard
1[Sat Feb 23 14:00:10 2019]
rule picard:
input: star_2pass/HB-9-2Aligned.out.sam
output: star_2pass/HB-9-2_rg_added_sorted.bam
jobid: 0
wildcards: sample=HB-9-2INFO 2019-02-23 14:00:12 AddOrReplaceReadGroups
** NOTE: Picard’s command line syntax is changing.
** For more information, please see:
** https://github.com/broadinstitute/picard/wiki/Command-Line-Syntax-Transition-For-Users-(Pre-Transition)
** The command line looks like this in the new syntax:
** AddOrReplaceReadGroups -I star_2pass/HB-9-2Aligned.out.sam -O star_2pass/HB-9-2_rg_added_sorted.bam -SO coordinate -RGID HB-9-2 -RGLB rna -RGPL illumina -RGPU hiseq -RGSM HB-9-2
14:00:12.937 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/home02/qizhengyang/anaconda3/share/picard-2.18.26-0/picard.jar!/com/intel/gkl/native/libgkl_compression.so
[Sat Feb 23 14:00:13 CST 2019] AddOrReplaceReadGroups INPUT=star_2pass/HB-9-2Aligned.out.sam OUTPUT=star_2pass/HB-9-2_rg_added_sorted.bam SORT_ORDER=coordinate RGID=HB-9-2 RGLB=rna RGPL=illumina RGPU=hiseq RGSM=HB-9-2 VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json USE_JDK_DEFLATER=false USE_JDK_INFLATER=false
[Sat Feb 23 14:00:13 CST 2019] Executing as qizhengyang@node21 on Linux 2.6.32-431.el6.x86_64 amd64; OpenJDK 64-Bit Server VM 1.8.0_121-b15; Deflater: Intel; Inflater: Intel; Provider GCS is not available; Picard version: 2.18.26-SNAPSHOT
INFO 2019-02-23 14:00:20 AddOrReplaceReadGroups Created read-group ID=HB-9-2 PL=illumina LB=rna SM=HB-9-2INFO 2019-02-23 14:00:58 AddOrReplaceReadGroups Processed 1,000,000 records. Elapsed time: 00:00:37s. Time for last 1,000,000: 37s. Last read position: chr1:10,583,000
[Sat Feb 23 14:07:47 CST 2019] picard.sam.AddOrReplaceReadGroups done. Elapsed time: 7.58 minutes.
Runtime.totalMemory()=999292928
To get help, see http://broadinstitute.github.io/picard/index.html#GettingHelp
Exception in thread “main” java.lang.OutOfMemoryError: GC overhead limit exceeded
at java.util.Arrays.copyOfRange(Arrays.java:3664)
at java.lang.String.(String.java:207)
at java.io.BufferedReader.readLine(BufferedReader.java:356)
at java.io.LineNumberReader.readLine(LineNumberReader.java:201)
at htsjdk.samtools.util.BufferedLineReader.readLine(BufferedLineReader.java:68)
at htsjdk.samtools.SAMTextReader.advanceLine(SAMTextReader.java:221)
at htsjdk.samtools.SAMTextReader.access$800(SAMTextReader.java:37)
at htsjdk.samtools.SAMTextReader$RecordIterator.next(SAMTextReader.java:257)
at htsjdk.samtools.SAMTextReader$RecordIterator.next(SAMTextReader.java:228)
at htsjdk.samtools.SamReader$AssertingIterator.next(SamReader.java:569)
at htsjdk.samtools.SamReader$AssertingIterator.next(SamReader.java:548)
at picard.sam.AddOrReplaceReadGroups.doWork(AddOrReplaceReadGroups.java:182)
at picard.cmdline.CommandLineProgram.instanceMain(CommandLineProgram.java:295)
at picard.cmdline.PicardCommandLine.instanceMain(PicardCommandLine.java:103)
at picard.cmdline.PicardCommandLine.main(PicardCommandLine.java:113)
Exception in thread “Thread-1” java.lang.OutOfMemoryError: GC overhead limit exceeded
at sun.nio.fs.UnixFileAttributes.get(UnixFileAttributes.java:68)
at sun.nio.fs.UnixFileSystemProvider.implDelete(UnixFileSystemProvider.java:227)
at sun.nio.fs.AbstractFileSystemProvider.delete(AbstractFileSystemProvider.java:103)
at java.nio.file.Files.delete(Files.java:1126)
at htsjdk.samtools.util.nio.DeleteOnExitPathHook.runHooks(DeleteOnExitPathHook.java:57)
at htsjdk.samtools.util.nio.DeleteOnExitPathHook$$Lambda$34/780934299.run(Unknown Source)
at java.lang.Thread.run(Thread.java:745)
[Sat Feb 23 14:09:07 2019]
Error in rule picard:
jobid: 0
output: star_2pass/HB-9-2_rg_added_sorted.bamRuleException:
CalledProcessError in line 115 of /home02/qizhengyang/qizhengyang/gatk_rna/Snakefile:
Command ‘set -euo pipefail; picard AddOrReplaceReadGroups I=star_2pass/HB-9-2Aligned.out.sam O=star_2pass/HB-9-2_rg_added_sorted.bam SO=coordinate RGID=HB-9-2 RGLB=rna RGPL=illumina RGPU=hiseq RGSM=HB-9-2’ returned non-zero exit status 1.
File “/home02/qizhengyang/qizhengyang/gatk_rna/Snakefile”, line 115, in __rule_picard
File “/home02/qizhengyang/anaconda3/lib/python3.6/concurrent/futures/thread.py”, line 56, in run
Removing output files of failed job picard since they might be corrupted:
star_2pass/HB-9-2_rg_added_sorted.bam
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
(END)
[qizhengyang@node1 gatk_rna]$ snakemake -n –quiet
Job counts:
count jobs
1 all
36 gatk
36 gatk_filter
16 gatk_split
1 picard
13 picard_markduplicat
1 | snakemake --unlock |
解决办法
查看 picard.jar 的路径
设置环境变量 export picard=/home02/qizhengyang/anaconda3/share/picard-2.18.26-0/picard.jar,
写入 ~/.bash_profile,source
设置JVM最大可用内存为40g,java -Xmx40g -jar $picard。默认是1G
[qizhengyang@node1 gatk_rna]$ find ~ -name picard.jar
/home02/qizhengyang/anaconda3/share/picard-2.18.26-0/picard.jar
/home02/qizhengyang/anaconda3/pkgs/picard-2.18.26-0/share/picard-2.18.26-0/picard.jar
查看当前环境变量 export | less
980879.node1 …b.gatk.202.sh qizhengyang 01:31:52 R low
980880.node1 …b.gatk.182.sh qizhengyang 01:31:40 R low
980881.node1 …b.gatk.204.sh qizhengyang 01:31:11 R low
980882.node1 …b.gatk.209.sh qizhengyang 01:31:20 R low
980884.node1 …b.gatk.214.sh qizhengyang 01:31:59 R low
980888.node1 …b.gatk.187.sh qizhengyang 01:30:53 R low
980889.node1 …b.gatk.210.sh qizhengyang 01:30:29 R low
980891.node1 …b.gatk.186.sh qizhengyang 01:31:41 R low
980892.node1 …b.gatk.216.sh qizhengyang 01:30:55 R low
980893.node1 …b.gatk.206.sh qizhengyang 01:31:36 R low
980894.node1 …b.gatk.192.sh qizhengyang 01:30:59 R low
980896.node1 …b.gatk.188.sh qizhengyang 01:31:45 R low
980898.node1 …b.gatk.215.sh qizhengyang 01:31:05 R low
980899.node1 …_split.167.sh qizhengyang 06:39:52 R low
980900.node1 …b.gatk.200.sh qizhengyang 01:31:05 R low
980901.node1 …_split.153.sh qizhengyang 05:35:11 R low
980902.node1 …b.gatk.201.sh qizhengyang 01:31:57 R low
980903.node1 …b.gatk.195.sh qizhengyang 01:29:26 R low
980904.node1 …b.gatk.197.sh qizhengyang 01:29:35 R low
980905.node1 …b.gatk.185.sh qizhengyang 01:30:31 R low
980909.node1 …_split.160.sh qizhengyang 05:36:38 R low
980910.node1 …b.gatk.198.sh qizhengyang 01:29:47 R low
980912.node1 …b.gatk.193.sh qizhengyang 01:30:22 R low
980913.node1 …_split.155.sh qizhengyang 04:15:22 R low
980914.node1 …_split.175.sh qizhengyang 04:04:21 R low
980915.node1 …_split.172.sh qizhengyang 03:43:49 R low
980916.node1 …_split.177.sh qizhengyang 03:22:15 R low
980917.node1 …_split.147.sh qizhengyang 05:08:37 R low
980918.node1 …_split.158.sh qizhengyang 05:14:32 R low
980919.node1 …_split.176.sh qizhengyang 04:49:57 R low
980920.node1 …_split.163.sh qizhengyang 04:37:11 R low
980921.node1 …_split.171.sh qizhengyang 04:17:19 R low
980922.node1 …_split.154.sh qizhengyang 04:05:50 R low
980923.node1 …_split.148.sh qizhengyang 03:46:37 R low
980925.node1 …_split.181.sh qizhengyang 03:25:10 R low
980926.node1 …_split.169.sh qizhengyang 02:20:52 R low
任务完成
[Sun Feb 24 23:55:12 2019]
Finished job 0.
103 of 103 steps (100%) done
Complete log: /home02/qizhengyang/qizhengyang/gatk_rna/.snakemake/log/2019-02-23T214006.459737.snakemake.log
最终的Snakefile
1 | # 2019-2-19 10:28:52 part |